
Fibrotic Pathways in IPF May Have a Common Protein and New Treatment Target
Written by |
A motor protein called myosin II was identified in a recent study to be a driver of fibrosis development — a finding with a notable potential to lead to improved treatments for idiopathic pulmonary fibrosis (IPF).
The study by Cleveland Clinic researchers is titled “Matrix-Driven Myosin II Mediates the Pro-Fibrotic Fibroblast Phenotype,“ and was published in the Journal of Biological Chemistry.
The type of myosin explored in the study — myosin II — is present in non-muscle cells. Myosins are involved in all sorts of cellular activities involving force, and myosin II is necessary for the contractions allowing fibroblasts, a key cell type in fibrosis development, to migrate and develop into more specialized cell types.
Earlier research showed that fibrosis development can be driven by three separate pathways, all of them acting upstream of myosin II. The lack of efficacy in lung fibrosis treatment is due, in part, to most treatments targeting one of these pathways. The activity in the remaining pathways is thought to still contribute to fibrosis and render treatments suboptimal. Myosin II in fibroblasts is located downstream of all these pathways, holding a strategic position where the three pathways converge.
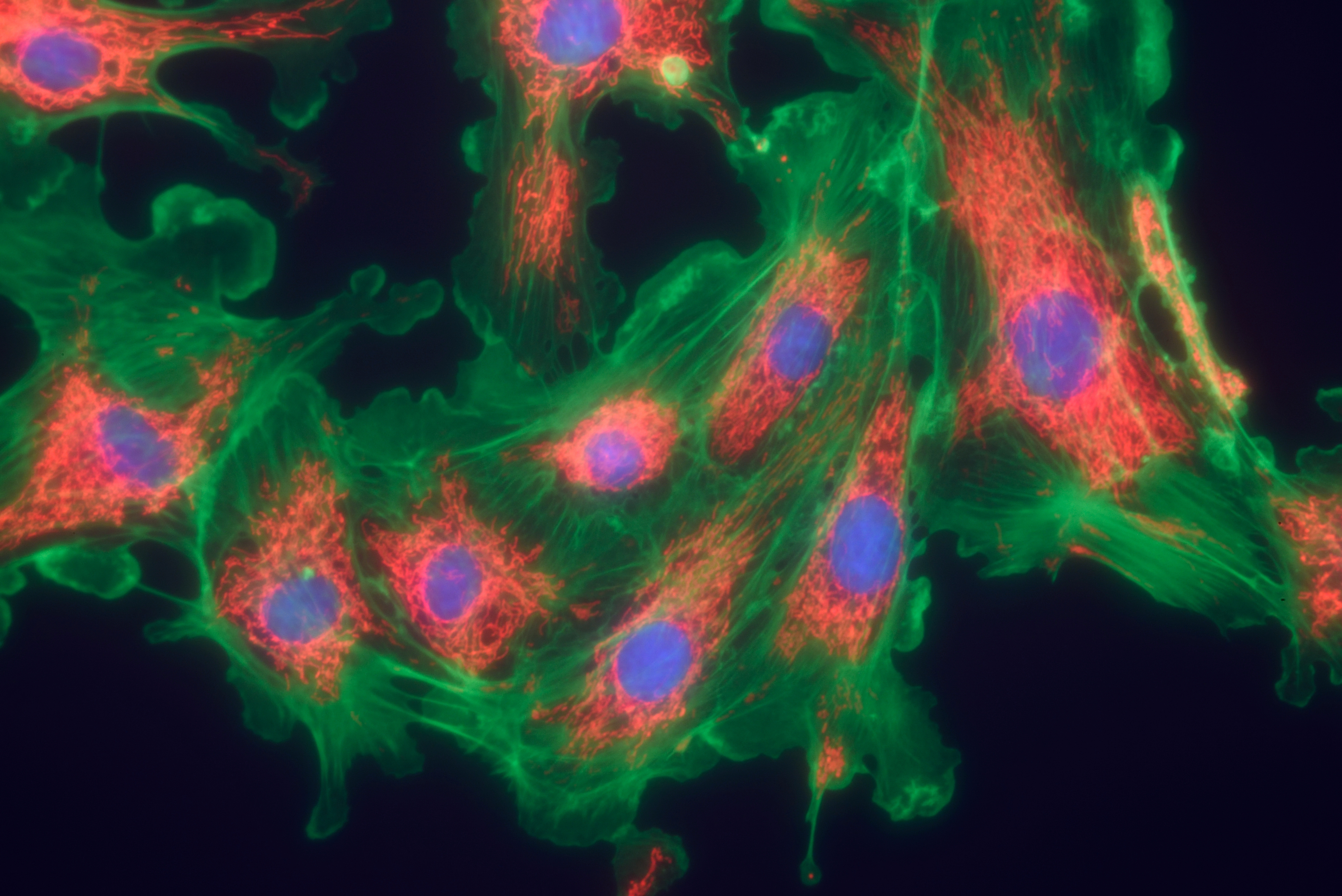
Led by Brian D. Southern, the research team used a new assay to study how fibroblasts are affected by different lung conditions. They isolated lung tissue from normal mice and mice with induced lung fibrosis, and allowed human fibroblasts to attach to the tissue. The team then tracked the cells using time-lapse video microscopy, noticing that fibroblasts growing on healthy tissue both looked and behaved differently than fibroblasts on fibrotic tissue.
On healthy tissue, fibroblasts were more motile and had a more polarized shape, while fibrotic tissue made the fibroblasts immobile with a more protrusive shape. Also, the cells growing on fibrotic tissue became more myofibroblast-like, with higher levels of the protein α-SMA — typical of fibrotic development.
Surprisingly, the group found that these changes were mediated by an increase in the activity of myosin II, and when the team inactivated the protein, the cells’ appearance and behavior returned to normal.
Researchers wondered if the stiffness of the diseased lung tissue is the signal driving these different fibroblast characteristics via myosin II signaling. To test this idea, they attached fibroblasts onto gels with stiffness mimicking normal (soft) and fibrotic (stiff) lung tissue, and studied the effects of myosin inactivation.
Results clearly showed that tissue stiffness drives fibrosis through myosin II. Blocking myosin II selectively influenced fibroblasts on the stiff matrix, preventing the development of myofibroblasts.
The authors believe that targeting myosin II could be an effective way to halt lung fibrosis, as well as fibrosis in other organs. “Future studies will focus on understanding this signaling through myosin II in more detail, and specifically in IPF (by using fibroblasts and lung tissue from actual patients with IPF). We will also start to look at ways to manipulate myosin II signaling as a potential treatment for fibrotic disorders,” the authors said.




Leave a comment
Fill in the required fields to post. Your email address will not be published.