Researchers Report New Understanding of Lung Fibrosis Physiopathology
Written by |
A new study offering insights into pulmonary fibrosis, entitled “Secreted frizzled related proteins inhibit fibrosis in vitro but appear redundant in vivo” was published in Fibrogenesis &Tissue Repair by Dr. Rik JU Lories’ research group at University Hospitals Leuven in Belgium. In this new research, the authors investigated the role of two extracellular Wnt antagonists, secreted frizzled-related protein-1 (SFRP1) and frizzled-related protein (FRZB) in lung fibrosis using in vitro and in vivo models.
The physiopathology of lung fibrosis is not clear, but it is suggested that the disease is caused by an abnormal repair response, and the disease is associated with significant morbidity and mortality, regardless of the disease’s origin in a patient. It seems that the activation of signaling pathways that are crucial during embryonic lung development may play a key role in the development of lung fibrosis. There is strong evidence supporting a central role for the activation of wingless-type antagonists like Wnt and transforming growth factor beta (TGFb) signaling pathways in the pathogenesis of lung fibrosis. Wnt ligands are lipid-modified, cysteine-rich glycoproteins that bind to frizzled (Fz) receptors. Wnt pathway activity is strongly regulated at the extracellular level by the secreted frizzled-related proteins (SFRPs), very similar to Fz receptors, which bind Wnt ligands in the extracellular space, thereby hypothetically preventing the binding of the ligand to the receptor.
[adrotate group=”3″]
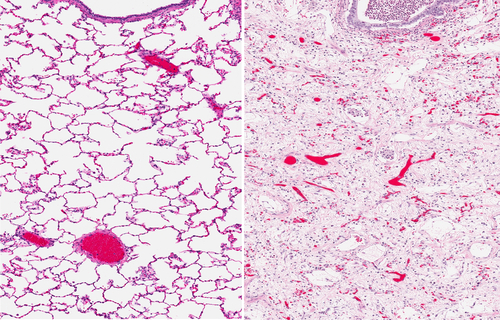
In this study, Dr. Ellen De Langhe and colleagues assessed the potential role of SFRP1 and FRZB in the development of lung fibrosis. For this, they used the TGFb-stimulated pulmonary fibroblasts and epithelial cells, as in vitro models, and the bleomycin-induced lung fibrosis model, as in vivo model, including the effect due to the absence of endogenous SFRP1 and FRZB, thus using mice lacking one of these proteins, respectively, Sfrp1−/− and Frzb−/− mice. They showed that both Sfrp1 and Frzb are more expressed during the course of bleomycin-induced lung fibrosis. In the in vitro studies, SFRP1 reduces the TGFβ1-induced increase of collagen expression in both pulmonary fibroblasts and alveolar epithelial cells. Using the mice deficient for Sfrp1 and Frzb, the Sfrp1−/− and Frzb−/− mice, the absence of either SFRP1 or FRZB did not alter fibrotic processes in the lung, suggesting that these proteins compensate functionally each other when one is absent.
The value of research such as this for patients with lung fibrosis lies in an increasing understanding of the disease’s physiopathology, which can in turn lead to improved therapies for treating not only the symptoms, but also the underlying causes.