Clusterin Protein in Lungs Can Both Protect Against and Promote Fibrosis, Study Shows
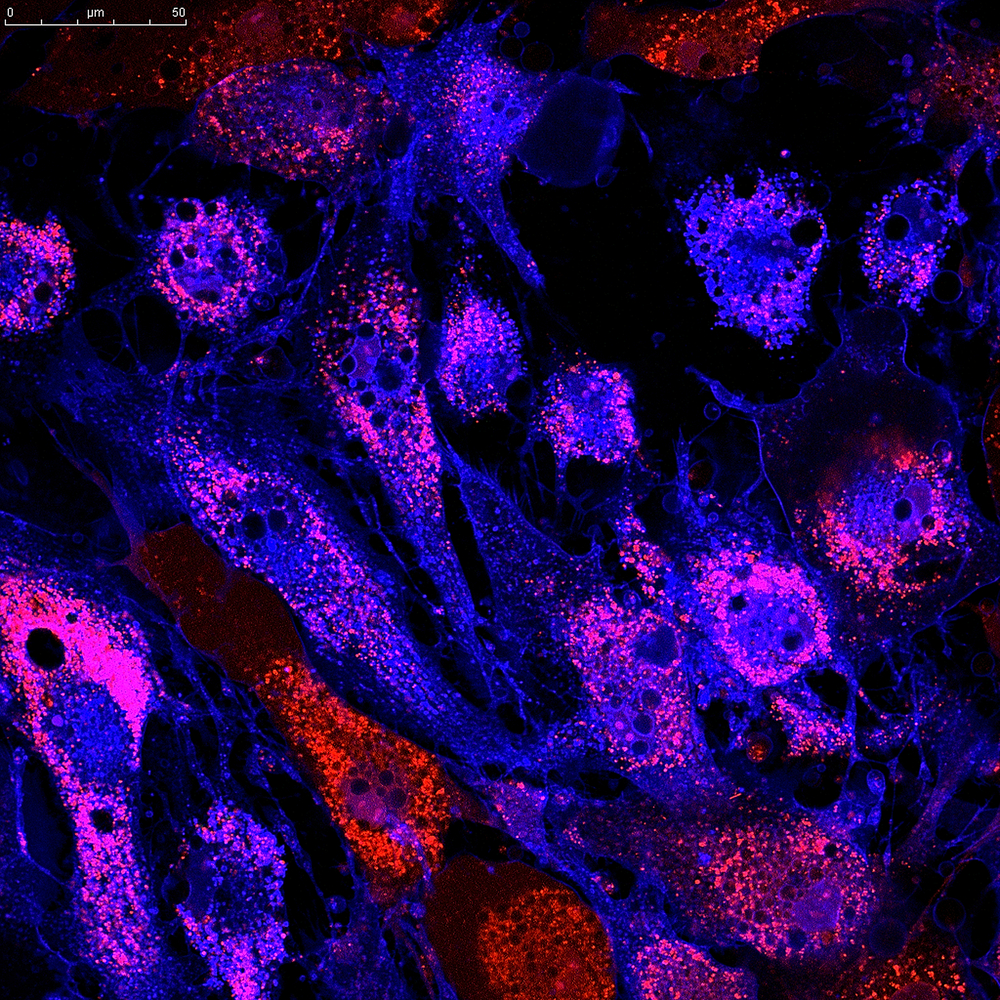



Cells’ attempts to limit fibroblast proliferation by lowering the levels of a protein called clusterin in lung fibroblasts of pulmonary fibrosis patients were found to be an insufficient strategy against fibrosis development in a recent study.
Researchers, in fact, observed that decreasing clusterin leads to a deposition of extracellular matrix proteins, which then actually contributes to fibrosis.
The study, “Diverse functions of clusterin promote and protect against the development of pulmonary fibrosis,” was published in the journal Scientific Reports.
Clusterin (also known as apoliprotein J) is present throughout the human body’s organs and tissues. In bronchoalveolar lavage fluid (BALF) samples from the lungs of idiopathic pulmonary fibrosis (IPF) patients, however, researchers found that this protein’s levels were markedly reduced — a decrease of up to sevenfold compared with healthy lungs.
Previous studies have linked clusterin levels with fibrosis progression, namely in the heart, kidney, and liver. In fact, the potent pro-fibrotic marker called transforming growth factor beta (TGF-β) has been shown to increase the levels of clusterin in epithelial cells.
In this study, researchers further investigated the mechanisms contributing to the decrease of clusterin levels in IPF BALF and its role in the development of pulmonary fibrosis.
They found that clusterin was strongly associated with normal fibroblast cells in healthy lungs, but its detection was weak or almost nonexistent in fibrotic regions.
Contrary to what was observed in epithelial cells, treatment of healthy lung fibroblasts with TGF-β reduced clusterin expression in a time-dependent manner.
Further experiments suggest that the intracellular levels of clusterin promote fibroblast proliferation, and subsequently fibrosis, but the same was not seen with clusterin provided exogenously, or outside the cell. Researchers found that exogenous clusterin actually protects fibroblasts from cell death.
“Our in vitro studies suggest that clusterin is protective against apoptosis [cell death] in normal and fibrotic lung fibroblasts in vitro,” the researchers wrote.
These results suggest that a decrease of clusterin in “IPF fibroblasts may be a physiologically appropriate, but insufficient, response of these cells intended to limit the development of an environment favoring unopposed fibroproliferation,” they said.
Additionally, the team found that the low levels of clusterin in IPF may contribute to an abnormal deposition of extracellular matrix proteins, which can lead to fibrosis.
Further studies are needed to shed light on the functional role of clusterin and its potential contribution to pulmonary fibrosis development.



